
脓毒症的新治疗策略


翻译:王剑荣 校对:王剑荣
摘要
在这篇小综述中,我们描述了多微生物导致脓毒症的分子机制,即一系列不良反应包括炎症和血栓前途径的激活、免疫系统缺陷和多器官功能障碍。补体激活是脓毒症的一个公认特征,尤其是C5a和C5b-9的产生,以及C5a相关受体的参与。C5a激活中性粒细胞导致DNA被挤出,形成含有髓过氧化物酶、氧化酶、以及细胞外组蛋白的中性粒细胞胞外诱捕器。产生下游补体激活产物,C5b-9(称为膜攻击复合物MAC)也在脓毒症中产生。C5b-9激活NLRP3炎症小体,损伤线粒体,同时在血浆中出现IL-1和IL-18。组蛋白具有强烈的促发炎和促血栓形成作用,导致血小板聚集与静脉血栓形成。多器官功能障碍也是脓毒症的一个特征。众所周知,脓毒性心肌病,如果严重,可以导致死亡。脓毒症的这种并发症与三种关键蛋白(SERCA2,NCX,Na+/K+-ATP酶)有关。这三种关键蛋白的减少是补体和组蛋白依赖性的。这些ATP酶的功能障碍与脓毒症的心肌病有关。这些数据可能提示人类脓毒症的理想的新靶点。
关键词
补体受体,补体,组蛋白,NLRP3炎症小体
简介
在北美感染性的脓毒症每年发病50多万人,死亡率为~40%,具体取决于脓毒症的严重程度。这些感染主要是由细菌引起的,但病毒、真菌和原生动物也可能引起脓毒症。尽管对动物和人类的脓毒症进行了广泛研究,尚没有一种联邦药品管理局(FDA)特别批准的药物专门治疗脓毒症。支持性治疗如液体复苏、广谱抗生素的早期应用和机械通气的安全技术已经改善了脓毒症患者的临床疗效。这个小综述描述了从小鼠多菌性脓毒症的研究获得的新见解或许可以给未来人类脓毒症研究铺平道路。
在人类和小鼠身上重要的基因组研究影响了我们对脓毒症的理解。几年前,一个研究小组利用了经过实验性脓毒症、烧伤等“炎性应激”的人类和小鼠的白细胞,进行研究了比较基因组学分析,Seok等人的报告得出结论:“小鼠的基因组反应模型很难模拟人类的炎症性疾病”。这导致很多报纸上的报道,暗示用小鼠与了解人类脓毒症相关性不大。2015年,日本的一个团队使用同样的数据库得出了相反的结论“小鼠模型中的基因组反应非常类似于人类炎症疾病”。截止到2018年,这一争议尚未解决,这代表了这一领域被困扰的问题,尤其是因为这两篇文章都发表在受人尊敬的科学杂志上(美国国家科学院院刊)。到现在为止对脓毒症小鼠的基因组分析能否可靠地推导到有脓毒症患者上没有共识,这导致了各大公司都避免在脓毒症领域进行研究投资,不管这些项目涉及多菌脓毒症的小鼠或人类的脓毒症。另一方面,应强调使用脓毒症小鼠已经允许在人类脓毒症中做不到的研究(例如测量心肌细胞中的调节蛋白,CMs)。脓毒症小鼠在信号传导方面路径方面以及如何操纵这些路径以减少脓毒症的器官功能障碍方面提供了重要信息(至少在小鼠身上)。最后,研究表明C5a补体产物与脓毒症小鼠和人类的脓毒症不良反应有关。小鼠基因敲除(KO)(缺乏C5a受体;C5aRs)已被证明可以预防在脓毒症的心肌病发展。这强调了在脓毒症小鼠中可以进行某些操作,提供重要信息关于脓毒症如何引起器官功能障碍这些资料可应用于感染的人类。
多微生物脓毒症中补体的作用
是多微生物性脓毒症常被用于大鼠和小鼠的脓毒症研究中,尤其是盲肠结扎和穿孔(CLP)引起的脓毒症,通常出现人类的临床表现非常相似脓毒症。该技术最初近40年以前被报道,目前已经被广泛使用,部分原因也是KO小鼠的存在。这项技术包括多次盲肠穿刺,把少量粪便挤出到腹腔。这个脓毒症的严重程度可由穿刺次数和针的大小(规格)决定。“多微生物”指存在脓毒症小鼠的腹腔和血液中出现需氧和厌氧细菌。多微生物脓毒症模型很少用于大型动物或类人灵长类动物是因为:在这种情况下,必须有一个重症监护室,需要24小时的动物护理,所有这些都会导致异常高昂的手术费用。此外,获得机构批准因为在非人灵长类动物身上使用CLP会有很大的问题。
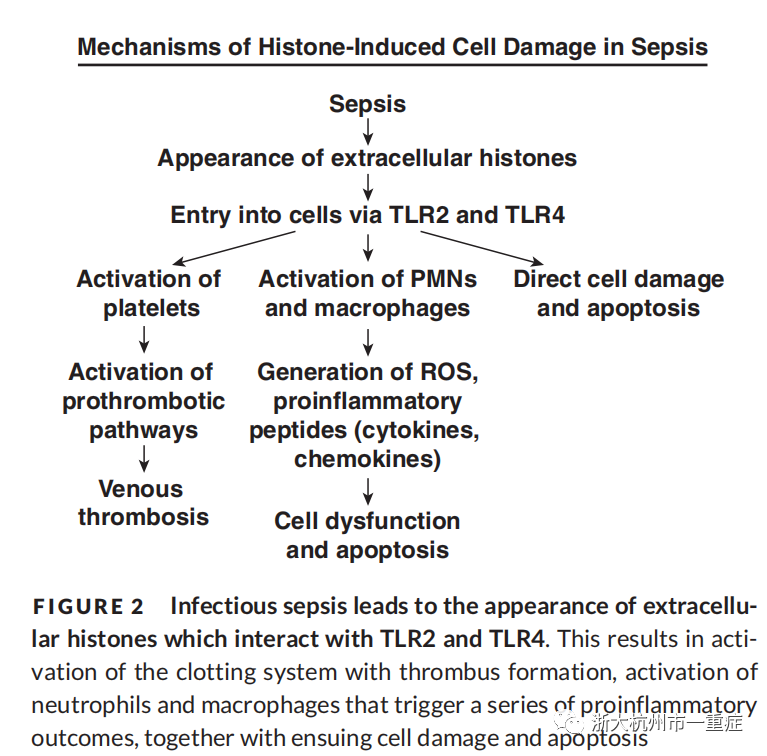
补体激活产物及相关受体参与多微生物脓毒症如图1所示。脓毒症的发生触发激活了几种补体途径,其中“经典”和“替代”途径在多微生物脓毒症中被激活。究竟是什么触发了这些途径的激活尚待确定。经典途径中的C5转化酶涉及C4b和C2a,而在替代路径C3b和Bb中代替了C5转化酶。无论哪种情况,都会生成C5a和C5b。目前尚不清楚是什么触发了传染性疾病的补体激活多微生物性脓毒症小鼠或人的脓毒症血症。目前公认的是革兰氏阴性菌通常通过凝集素途径相互作用激活补体。其特征是甘露糖凝集素在细菌表面与碳水化合物结合。与先天免疫系统有关,有很多模式相关的模式分子和损伤相关的模式分子,可以激活补体。例如,细菌革兰阴性菌LPS是一种著名的细菌产物可以激活补体替代途径。脂多糖与TLR2和TLR4相互作用可激活多种细胞(图2)。最后,某些革兰氏阳性细菌(金黄色葡萄球菌)可以分泌阻止补体分子激活的蛋白质。第三途径(“凝集素”途径)有时是细菌引起的补体激活的主要途径。无论什么途径被触发,都有C5a的出现,这种强大的过敏毒素与它的受体(C5AR1和C5AR2)发生反应。尤其在中性粒细胞(PMN)中普遍存在。结果

图1感染性脓毒症发生的事件顺序:补体激活的重要作用产生两大产物,C5a和C5b-9(膜攻击)的产生复杂)。C5a激活多形核白细胞(中性粒细胞)形成网,释放细胞外组蛋白和促炎肽类。C5b-9激活NLRP3炎症小体,导致两种有效细胞因子IL-1和IL-18的释放。
有很多PMN强烈被激活,其次还有一些巨噬细胞被一定程度激活。激活的PMN释放长链DNA,形成中性粒细胞胞外陷阱(NETs),同时释放组蛋白以及PMN特异性颗粒(髓过氧化物酶、蛋白酶等)的产物。组蛋白(H1、H2A、H2B、H3、H4)是促炎和促血栓形成活性的有效生物反应诱导分子。如图2所示,细胞外组蛋白在人和小鼠脓毒症症中起关键作用效应器的作用。组蛋白可引起弥漫性血管和上皮细胞损伤,在脓毒症多器官功能障碍中起重要作用。下游补体激活也导致C5b-9(膜攻击复合物;MAC)的产生(图3)。
图2感染性脓毒症导致与TLR2和TLR4相互作用的细胞外组蛋白的出现。这导致血栓形成时凝血系统的激活,激活中性粒细胞和巨噬细胞触发一系列促炎症反应结果,以及随之而来的细胞损伤和凋亡。
重要的是要强调补体激活通道
经典途径或替代途径产生C5转化酶,产生C5a和C5b。C5a与C5aR在PMN中相互作用,导致产生NETs,释放组蛋白强大的促炎功能。C5b的生成与C6、C7、C8和C9形成C5b-9。我们知道这个复合体会激活NLRP3炎症小体导致炎症小体激活,线粒体损伤和IL-1和IL-18这些强大的促炎细胞因子的释放。它的NLRP3炎症小体前驱体模式存在于多种细胞(PMNs、巨噬细胞)中,心肌细胞、星形胶质细胞等)。
简而言之,炎症小体程序包括:“启动”阶段,细胞暴露于脂多糖(100 ng/mL)的在37°C下持续4小时。这导致少量IL-1β释放。此后,细胞暴露在1mmol/L的ATP中45分钟,ATP作为一种“激活剂”,导致大量释放IL-1β。Suresh等人。同时也证明了补体介导的“旁观者”损伤,在巨噬细胞吞噬补体调理颗粒过程中,引发NLRP3炎症小体活化,导致caspase-1活化并IL-1β、IL-18分泌,补体依赖性激活或炎性激活可通过上述机制引起急性细胞和器官损伤(通过C5a和C5b-9引起的功能障碍)。当补体或炎症激活发生时,就可能发生这种不良事件。从这个意义上讲,细胞损伤不会的发生是一种非靶向性的方式,在炎症发生在补体或炎性小体激活的附近,使多种细胞受到破坏。
关于细菌和补体系统,革兰氏阳性细菌,如金黄色葡萄球菌,目前比较确定的是革兰氏阳性菌耐受补体激活效应。而革兰阴性菌对补体激活活化的影响敏感。例如,金黄色葡萄球菌对甲氧西林耐药,可能会造成对病人严重的危险。还应强调的是,许多格兰阴性菌释放与TLRs(2、3、4和9)反应的LPS,在脓毒症的条件下可能共同导致感染性脓毒血症的事件。补体激活产物,如C3b和iC3b通过PMNs和巨噬细胞促进细菌吞噬和胞内杀伤,通常借助NADPH氧化酶及其产物、氧自由基。另外,如前所述,MAC对细菌还有溶细胞活性,一般通过MAC或凝集素和NLRP3炎症途径。细菌同样也能激活凝集素补体途径,产生如上所述的抗菌作用。还应注意的是,某些革兰氏阳性菌(如金黄色葡萄球菌)释放的肽类能阻断补体活化或补体活化产物的激活。
补体复合物如C5b-7或C5b-8有无细胞损伤活性在实验研究中基本上没有令人信服的依据。摩根最近的一项研究研究组发现,在没有C9的情况下,通过研究细胞因子产生、炎性小体激活、细胞溶质钙离子增加、线粒体功能障碍和细胞色素c释放情况发现:MAC亚体不能导致人肺部炎症和导致上皮细胞凋亡。应该提到的是大多数与C5b-7和C5b-8相关的报道均未提示其对哺乳动物细胞有损伤作用。
脓毒症病人组蛋白诱导的细胞损伤机制
脓毒症的细胞损伤图2描述了我们最近的工作,它定义了CLP诱导的脓毒症发病后心脏中发生的信号通路激活。主要的事件是C5a激活PMNs,导致NETs以及细胞外组蛋白出现。已经证实组蛋白与包括CMs的各种细胞类型的TLR结合。这导致血小板活化与血栓形成。另一个主要事件是在细胞外组蛋白存在激活PMN和巨噬细胞,导致各种细胞中产生氧化剂(活性氧ROS),同样也产生各种促炎肽(细胞因子、趋化因子、以及其他因素),所有这些都会导致细胞功能障碍,而且,通常导致凋亡。尤其是在感染性脓毒症的环境中。组蛋白的其他损伤特征是它们可以直接导致细胞损伤和细胞凋亡,并且具有促血栓和促炎症作用。很多研究表明,组蛋白与不同的TLR结合(主要是TLR2和TLR4)并与不同细胞类型上的受体相互作用,这取决于实验设置和使用的细胞系不同,有关于组蛋白激活TLR2或TLR4的额外数据。Xu等人的研究显示TLR4,但不是TLR2,是组蛋白诱导的细胞因子炎症的主要受体,根据TLR4 KO小鼠输注组蛋白后的反应研究。Ekaney等人还表明,阻断TLR4导致人内皮细胞的细胞毒性(根据乳酸脱氢酶测定和碘化丙啶染色)。它已经证明,TLR2和TLR4阻断性抗体阻断抑制了内皮细胞的细胞毒性和抗血管生成作用。我们还发现了来自双TLR2和TLR4 KO小鼠的巨噬细胞比单独TLR2或TLR4KO小鼠的巨噬细胞能显著减少暴露于组蛋白后炎症引起的IL-1β释放。
心脏的分子基础
在聚微生物脓毒症是图3描述了感染性脓毒症如何激活一系列对小鼠心脏功能不利影响的信号通路。最初,MAPK,特别是p38和Akt信号通路激活,随后血浆中各种细胞因子,趋化因子和细胞外组蛋白水平升高。这些事件的结果引起心肌细胞的严重功能障碍。心肌功能不全的基础与p38激活有关,因为水溶性p38抑制剂阻断脓毒症引起的心功能不全(如由超声多普勒参数确定)的发生。脓毒症心肌病的原因可追溯到Na+K+ATP酶的功能不全,这一点至关重要对于心肌细胞和其他细胞类型的有效动作电位。内质网Ca2+ATP酶 2型(SERCA2)和Na+/Ca2+(NCX)交换泵导致心肌舒张期[Ca2+]i积聚(以一种与心肌舒张期胞浆[Ca2+]i清除障碍相关的方式)。这些事件导致心肌细胞的实质性功能障碍和凋亡。我们相信这些事件可能同样发生在其他器官(脑、肝、肺等),导致脓毒症多器官功能障碍。根据这些信息,阻断p38的激活可能是一种预防脓毒症性心功能不全发生的策略。

图3感染性脓毒症引起脓毒症心肌病的事件,定义为心肌细胞收缩和舒张缺陷。结果可以追溯到心肌细胞中MAPK(p38)和Akt激活,导致心功能不全。这些缺陷与动作电位缺陷(Na+/K+-ATP酶降低),以及[Ca2+]i调节蛋白、SERCA2和Na+/Ca2+交换酶活性的降低有关。
结论
图中的数据。1-3强调脓毒症出现时补体的激活产物和组蛋白最终导致心脏功能障碍。因为C5a是诱发在感染性心肌病中发展的很多缺陷的关键因素,体内阻断C5a或者C5aRs对脓毒症小鼠有明显的潜在治疗作用。
这种抑制剂目前还没有,仍在研制中。在人类的严重脓毒症中,大多数致命性事件发生在败血症发病后的最初3-5天,尤其是老年脓毒症患者。C5a或信号途径抑制剂只在有限的时间内(3-5天)。目前,有几家制药公司正在开发能阻断C5a或C5的小分子化合物,尽管尚未进行临床试验以评估这些化合物安全性和的功效。就C5b-9阻断药而言除了C5抗体(结果是阻断C5b-9的产生),似乎还没有任何FDA批准的药物。但是,使用人C5(依库丽珠单抗eculizumab)单克隆抗体阻止C5a和C5b-9产生,也与脑膜炎球菌性脑膜炎的发生倾向有关。有一些影响补体系统的疾病,如阵发性夜间血红蛋白尿以及非典型溶血性尿毒症综合征,临床试验表明eculizumab是有效和安全的。然而,在感染性脓毒症中如果合并菌血症,使用抗C5很可能面临FDA批准的门槛很高,此类干预的安全性临床试验需要花费很高。从短期来看似乎不太可能获得FDA的批准。发展小分子抑制剂比阻断C9的活化表位是阻断C5b-9生物活性的一个有吸引力的策略。
目前有一些单抗可以阻断活化的C9的表位,但是这种单克隆抗体还没有用于人类。在感染性人类中使用复合IL-1受体激动剂(IL-1ra)靶向的IL-1(NLRP3炎症激活产生)的尝试已经失败,联合靶向IL-1、IL-18的药物治疗脓毒症小鼠的研究显示了有希望的数据。Vanden Berghe等人,使用CLP或LPS诱导的小鼠内毒素血症研究表明IL-1r拮抗剂和抗IL-18的中和作用对小鼠的内毒素致死性起到保护作用。他们也在应用IL-1和IL-18基因缺陷小鼠研究发现了类似的结果。尽管需要临床研究来确认小鼠的联合治疗数据,IL-1阻断的临床实验的失败阻止了加做阻断IL-1ra为特征的临床试验。有趣的是,除脓毒症外,阻断C5a或其受体或C5b-9可能还有与人类的许多其他疾病相关,如急性心脏病缺血、急性肺损伤、自身免疫性疾病(如类风湿性关节炎,系统性红斑狼疮)和早期移植排斥反应。

相关阅读
- 09-13 第八期“浙大市一.临床大讲堂”预告
- 05-24 2021年第三届湖畔眼底病高峰论坛暨浙江省级继续医学教育项目《玻璃体视网膜疾病诊疗进展》圆满举办
- 10-29 COVID-19流行期间重症监护中的床旁肺部超声
- 10-29 肝硬化患者的自发性细菌性腹膜炎和腹膜外感染(上)
- 10-20 妊高症回顾(上)
- 10-20 妊娠期高血压疾病与心血管疾病相关发病率和死亡率的系统评价分析
- 10-07 急性心梗后超声可发现的机械性并发症
- 10-07 做了一回赵半仙--高血压心脏病
- 10-07 综合生命支持降低暴发性心肌炎死亡率的多中心研究(上)
- 10-07 经鼻雾化吸入(上)