
脑卒中相关的肺损伤:从病理生理到临床实践


翻译:陈明月 编辑:孙雁鸣
Robba等人通过回顾性病例研究探索了急性缺血性卒中后脑-肺相互作用的病理生理学机制,以及机械通气的管理。急性缺血性脑卒中(急性脑梗死)后继发肺部并发症很常见,且死亡率很高,其机制被称为“双重打击模型”,这似乎足以解释某些病理生理现象。但事实上,该机制可能更为复杂。
急性细菌性呼吸道感染,尤其是发生于脑卒中前1周,可能是脑梗死的重要危险因素;急性感染表现为急性中风的诱因。因此,不能排除某些报告为脑卒中后早期感染的,实际上是脑卒中发作之前已经存在,而后进一步恶化。
我们同意,吞咽困难和意识障碍以及其他机制可能与脑卒中相关性肺炎有关。但应注意到,脑卒中后会发生免疫激活或免疫抑制;正常的脑-免疫相互作用可能会失调,且已得到实验和临床数据的支持。事实上,PREDICT研究证实,脑卒中诱发的免疫抑制综合征是脑卒中相关性肺炎的独立危险因素,而非误吸。
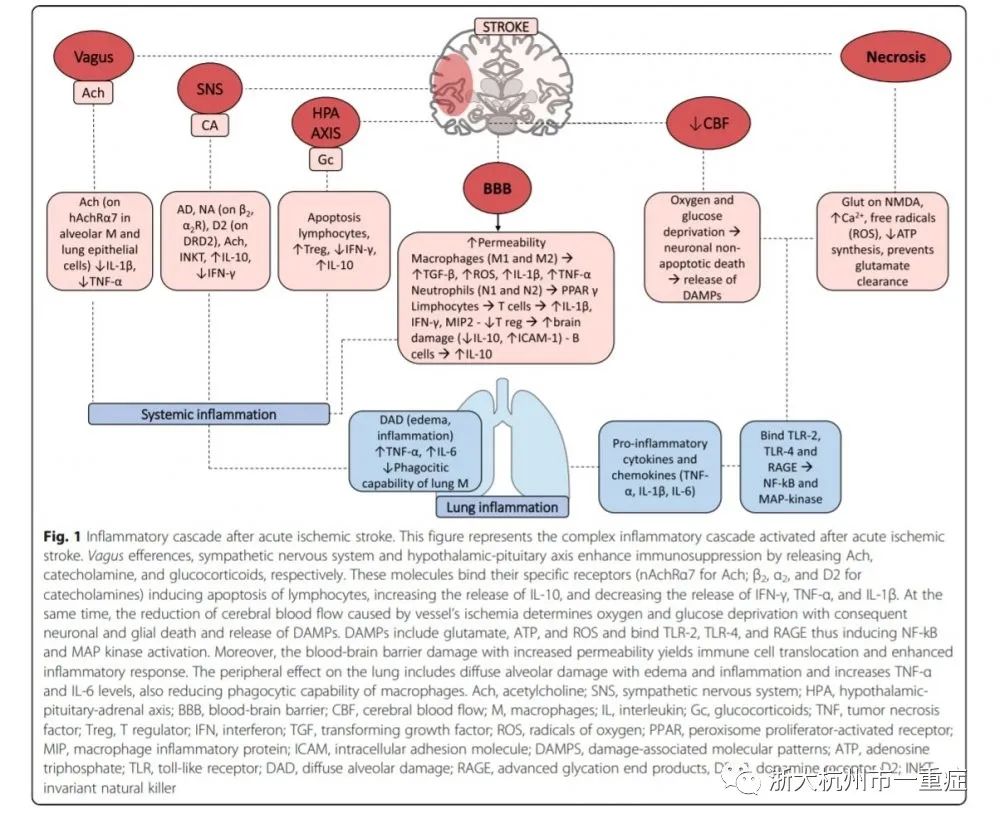
急性缺血性脑损伤会迅速激活交感、副交感和HPA(下丘脑-垂体-肾上腺)轴,导致去甲肾上腺素(NE),乙酰胆碱和糖皮质激素(GCs)的释放。肾上腺素能通路和HPA轴通路协同,通过NE和GCs的协同作用导致脾萎缩和自然杀伤细胞(NK)缺乏。此外,缺血性脑卒中小鼠的支气管肺泡灌洗液发现,巨噬细胞和中性粒细胞以及全肺组织促炎性IL-1β mRNA表达显着增加。目前已知IL-1β与急性肺损伤(ALI)和/或ARDS有关,且已被证明是ALI中生物活性最强的细胞因子之一。
综上,急性缺血性脑卒中患者的肺部损伤增加了死亡风险。脑损伤后肺部损伤的机制值得进一步研究。
事实上脑-肺相互作用机制很复杂,仍需进一步研究。如下图所示,我们展示了急性缺血性脑卒中诱发的肺部炎症的主要机制。
血管闭塞后,氧气和葡萄糖的缺乏会导致神经元和神经胶质细胞死亡,随后释放损伤相关分子模式(DAMPs),如ATP、谷氨酸和活性氧(ROS);这些分子与toll样受体(如TLRs-2、TLR-4)和晚期糖基化终产物(RAGE)受体结合,激活并刺激小胶质细胞释放促炎细胞因子(例如TNF-α,IL-1β和IL-6)。谷氨酸与N-甲基-D-天冬氨酸受体(NMDA)结合可增加钙内流,从而产生ROS。
急性缺血性脑卒中后,交感神经系统,传出迷走神经和HPA轴过度激活,从而可能引起免疫抑制。传出迷走神经释放乙酰胆碱(Ach),在与烟碱乙酰胆碱受体(nAchRα7)结合后,可削弱肺泡巨噬细胞和上皮细胞的炎症反应,并削弱神经细胞对氧化应激的抵抗力。交感神经系统释放儿茶酚胺(作用于β2、α2和D2受体),与干扰素(IFN)-γ和IL-10协同激活调节T淋巴细胞。此外,HPA轴释放糖皮质激素,从而促使淋巴细胞凋亡,促进释放IL-10,抑制T细胞释放IFN-γ。
血脑屏障通透性增加,发源于颅内的炎性因子释放到体循环中。小胶质细胞(M)极化为M1(经典激活)或M2(替代激活)2种亚型;M2型通过释放转化生长因子-β保护脑细胞,而炎性因子诱导的M1型可加剧脑梗死后脑损伤。
因此,急性缺血性脑卒中后全身性反应炎症被激活。在急性缺血性脑卒中小鼠中观察到弥漫性肺泡损伤(肺水肿和炎症损伤),总体与促炎细胞因子增强和肺巨噬细胞吞噬能力下降有关。
尽管在脑卒中的发病机制及其与其他器官的相互作用方面取得了重大进展,但仍需要进一步的研究来阐明脑卒中诱发的免疫抑制及其潜在的治疗机制。


相关阅读
- 09-13 第八期“浙大市一.临床大讲堂”预告
- 05-24 2021年第三届湖畔眼底病高峰论坛暨浙江省级继续医学教育项目《玻璃体视网膜疾病诊疗进展》圆满举办
- 10-29 COVID-19流行期间重症监护中的床旁肺部超声
- 10-29 肝硬化患者的自发性细菌性腹膜炎和腹膜外感染(上)
- 10-20 妊高症回顾(上)
- 10-20 妊娠期高血压疾病与心血管疾病相关发病率和死亡率的系统评价分析
- 10-07 急性心梗后超声可发现的机械性并发症
- 10-07 做了一回赵半仙--高血压心脏病
- 10-07 综合生命支持降低暴发性心肌炎死亡率的多中心研究(上)
- 10-07 经鼻雾化吸入(上)